Transcriptomics Sandbox¶



Operating System:

Shell:

Editor:



Package Manager:




Programming Language:




Operating System:

Shell:

Editor:



Package Manager:




Programming Language:




Operating System:
Shell:
Editor:
Package Manager:

Programming Language:


Operating System:
Shell:
Editor:
Package Manager:

Programming Language:

In this app you will find transcriptomics related courses you can learn from, and datasets and tools you can work with for your own research/learning purposes. The included materials and tools are organized by the Health Data Science sandbox.
The coding modules of this sandbox are currently based on Rstudio Server and JupyterLab. Rstudio Server and Jupyterlab are web-based integrated development environments for (and not limited to) R, Python and Bash programming languages. They can create interactive coding material (Rmarkdown and Jupyter notebooks), which includes text, animations, code, and data.
Available Items¶
Learning material and tools are periodically added to this sandbox and can be chosen from the menu. Each module can be a course, a setup to work with specific software, a research example, etc. Each module come with all necessary packages installed, eventual notebooks with computer code and explanations, and a dedicated webpage with additional material (notes, slides, recordings, ...) when this is available.
The available tools are:
Tool Name |
Description |
Links |
Programming Language |
|---|---|---|---|
RNAseq in Rstudio |
Rstudio session with common bulk and single cell RNAseq packages such as DESeq2, Seurat and clusterProfiler. |
- |
R |
RNAseq CLI |
JupyterLab session containing common transcriptomics tools (for single cell-RNA and bulk-RNA data analysis), ideal to create your own pipelines (Snakemake, Nextflow or GWF). It includes all packages from the Rstudio session as well. |
- |
Python, R |
Cirrocumulus |
Cirrocumulus is an interactive visualization tool for large-scale single-cell genomics data. |
Python, JavaScript |
|
Introduction to Bulk |
A 3-day course to introduce bulk RNAseq analysis, from data alignment to bioinformatics analysis. |
Bash, R |
|
Introduction to |
A 2-day course to introduce single cell RNAseq analysis in R. |
R |
|
Advanced Single Cell |
Research-based examples for advanced topics in single cell analysis. |
Bash, Python, R |
Note
Course materials will be automatically downloaded unless you have added a folder called Intro_to_bulkRNAseq, Intro_to_scRNAseq_R, or AdvancedSingleCell, respectively, in the submission step of the Transcriptomics app.
How to Use the Modules¶
Our modules are designed to be user-friendly and easy to use. Some are for self-learning and come from courses/workshop held in danish universities. Other are tools you can use for your own purpose. Here are some information to get you started:
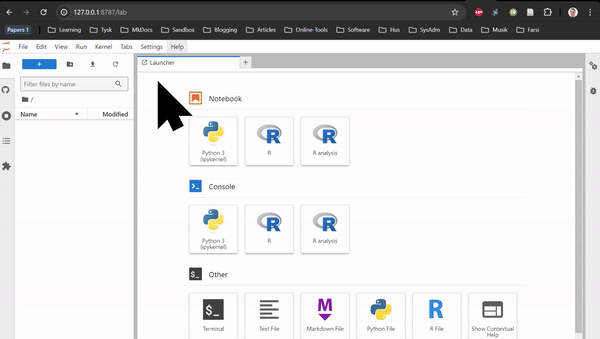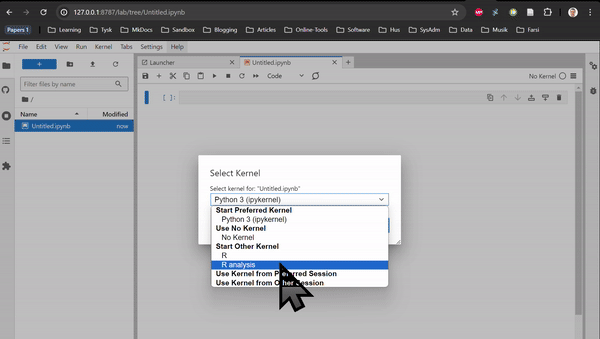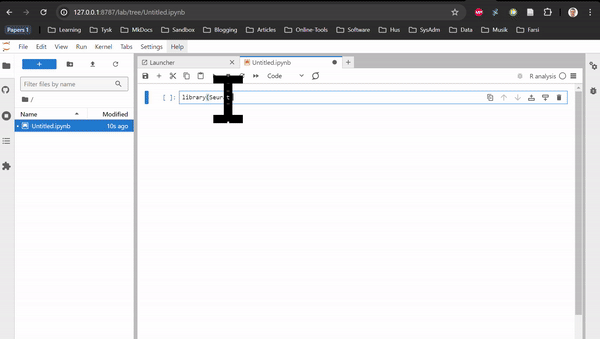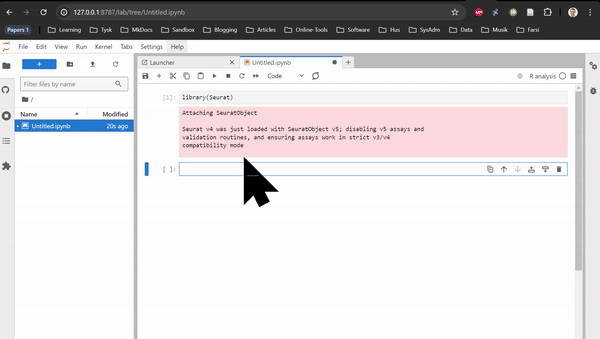
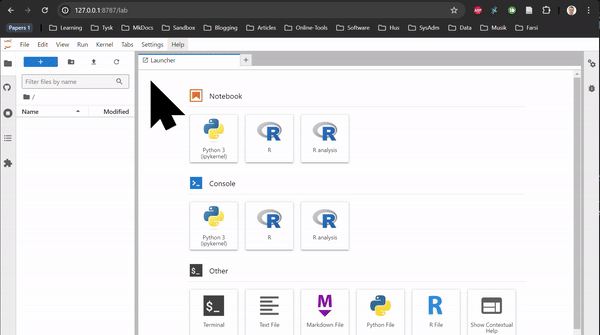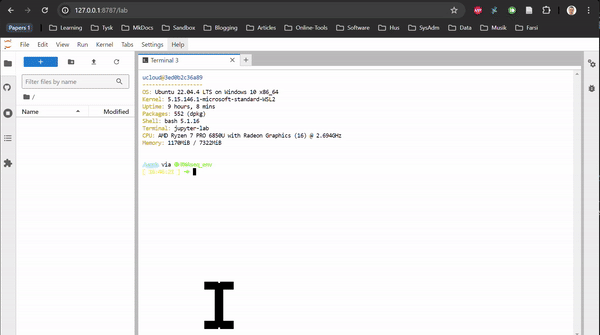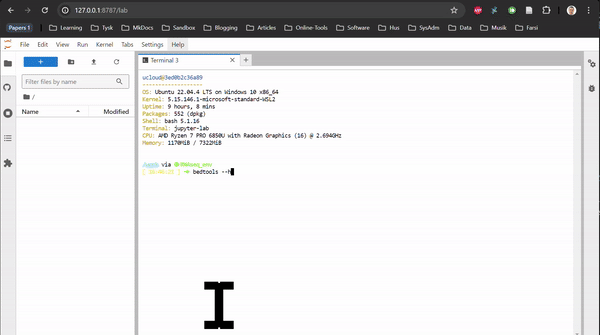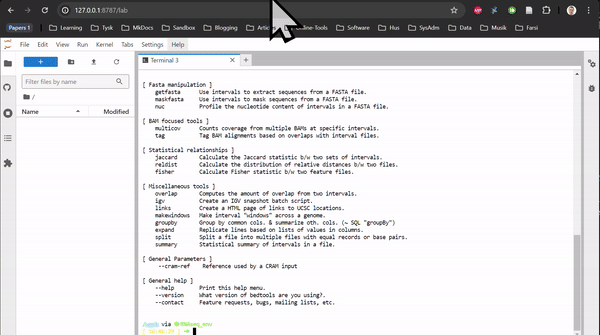
RNAseq in Rstudio and RNAseq CLI are based on Rstudio and JupyterLab, respectively. They do not contain any tutorial, just all the packages used in the tutorials of the Transcriptomics Sandbox. In JupyterLab, you can both use the command line in the terminal and the notebooks through the available kernels. See the two animations below for how you start a notebook or a terminal in JupyterLab.
Cirrocumulus is a tool to visualize single cell RNAseq data. You can explore datasets and visualize them in a user-friendly way. You can also print out gene expression plots, heatmaps and perform differential gene expression for selected cell groups.
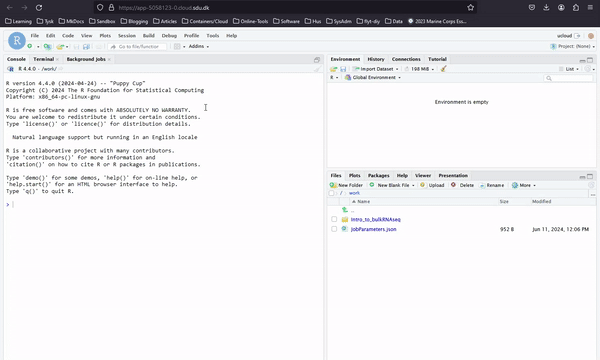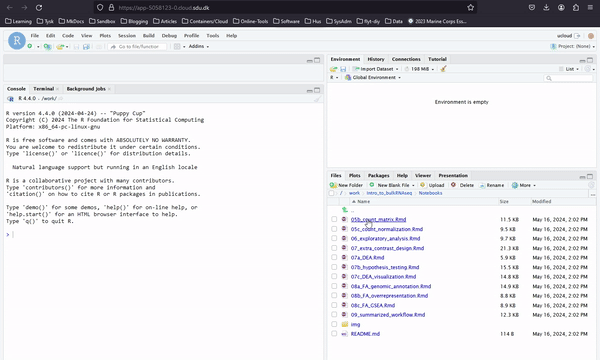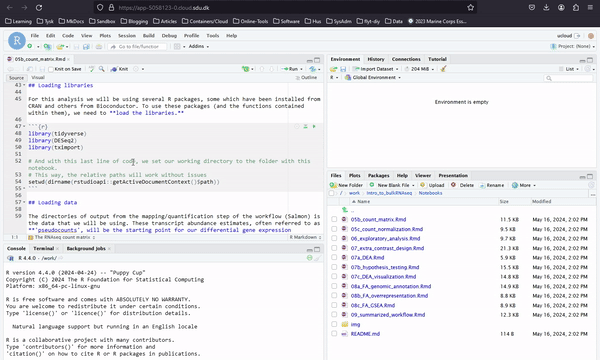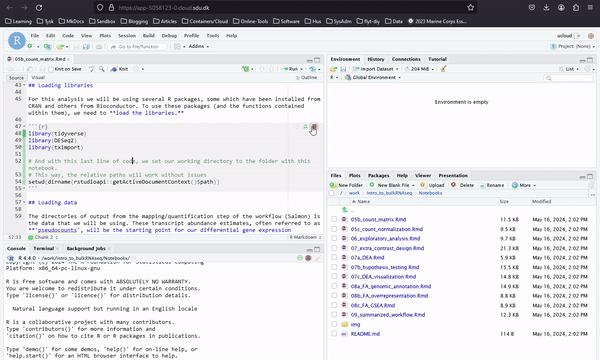
In Introduction to Bulk RNAseq Analysis you can access all the notebooks of an introductory bulk RNA analysis course, as shown below. Here you work with Rstudio. Slides and other materials are available through this webpage.
In Introduction to Single Cell RNAseq Analysis you can access all the notebooks of an introductory single cell RNA analysis course similarly to the bulk RNA course above. Here you work with Rstudio. Slides and other materials are available through this webpage
In Advanced Single Cell Analysis you can run various tutorials and examples of advanced single cell analysis. Here you work with JupyterLab and Jupyter notebooks. Slides and other materials are available through this webpage. Look below to see how to access the notebooks through the browser folder.
Hint
Make a copy of any notebook or R markdown, upload your own data, and replicate any analysis you wish on your own data by modifying the code we make available!
Save Your Work¶
Every time you launch a module, everything is saved in the default folder you are working on. This will be saved in your user folder after you finish your job in UCloud. The user folder is usually called Username#Number. This contains a folder called Jobs. Here you find all the software you executed, and for each execution the saved material which you can retrieve.
Retrieve Your Work¶
To retrieve your work from a previous session of a module, add a folder in the job submission page which is named Intro_to_bulkRNAseq, Intro_to_scRNAseq_R, or AdvancedSingleCell, depending on which module you need to run. See the example below for the Advanced Single Cell Analysis module.
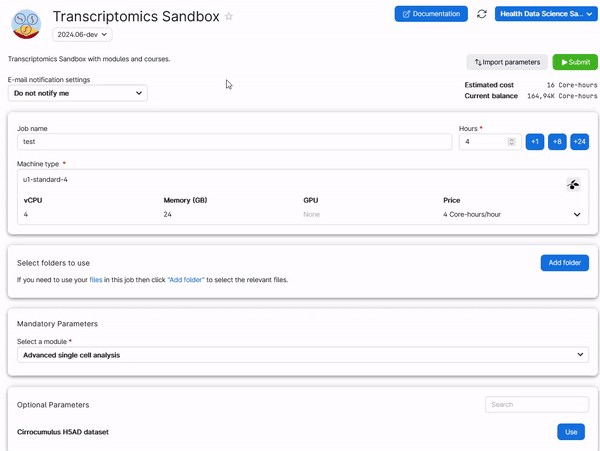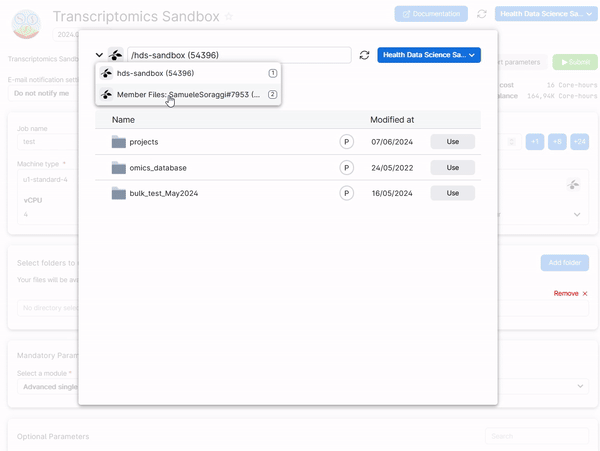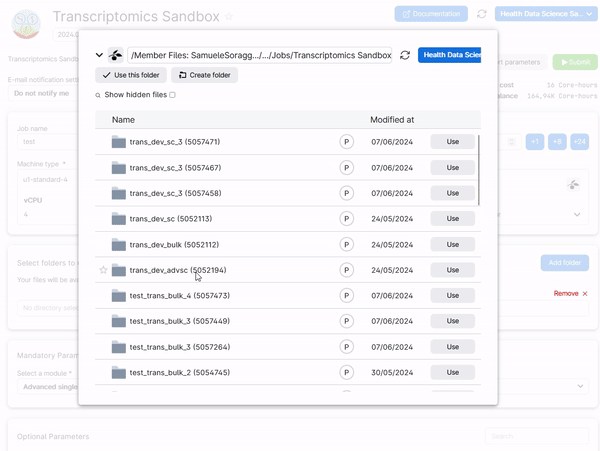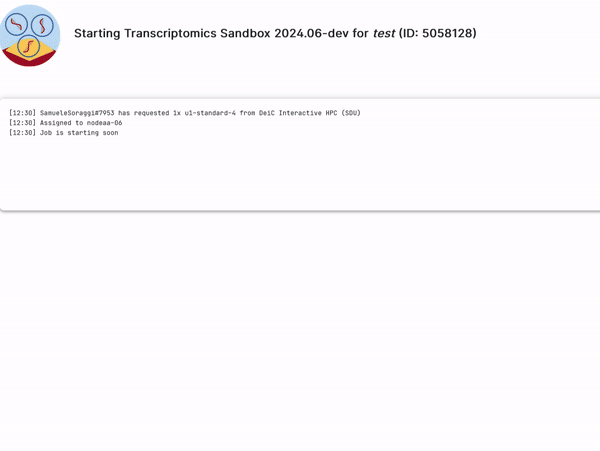
Hint
Only the folders you add in the submission step will be visible when using the app. If you upload data through the UCloud interface, remember to add the folder containing your data if you need to use a module on it.
Contents